What a Shapely genome you have!
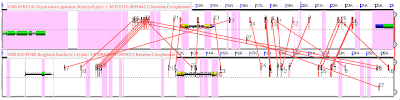
This might be a case of if you have a really cool hammer , everything looks like a nail, but it was fun mixing tools from different disciplines. After finding synteny , there's a bunch of paired genes whose neighbors are also pairs. Paired ( homologous ) genes have similar sequence because they have some function and can't change without loss of function. Non-gene sequence between the paired genes is mostly randomized via mutation, deletion, etc. But, there is non-gene sequence that is conserved between the genes. These CNS's-- conserved non-coding sequences--are usually sites that bind stuff that regulates the expression of a gene. That looks like this. With one gene on the top, and its pair below, both yellow. Pink lines in the foreground connect putative CNSs (similar sequences) between these genes. That the lines cross is bad. CNSs occur right at the level of noise. So even though a similar sequence occurs near both genes, it could be by chance. It is possible to red...