genome scrubber : mask repetitive sequence
This is to describe a simple tool I've made available (svn repo) for masking repetitive sequence.
rice (Oryza Sativa) version 5 sequence looks something like below when run through pyfasta info.
So, it's not huge (still only 1/10th the size of human) but, it can be difficult to deal with the entire genome because of the large amount of repetitive sequence and transposable elements. This is sometimes mistakenly referred to as "junk DNA", while that's not true, it does make whole-genome analyses a pain as a the output is dominated by repetitive sequences matching their own families. Doing a blast of the rice genome with this command:
nearly causes my machine to run out of memory (16G), takes a couple of days to run, and results in a blast output of 5.1G and 84 million rows--that's 84 million blast hits with an e-value below 0.001! By definition, that output is dominated by the repetitive elements. Repetitive elements are interesting, but in the case were we want to look at synteny, we have to wade through that 5.1G of stuff to find the very small chunk of data we need. This adds time to run the sequence comparison, time to parse, time to plot, time to analyze, and data to store, etc...
The solution we use in our lab is to create a "masked" sequence where we 'X' out any DNA sequence occurring more than a given number of times in the original blast. So, from the output of the blast above, any basepair that is covered more than 50 times is hard-masked with 'X'. The single script to run this is here. Available from svn via:
when run as a script with no options, it will print some help text. To mask rice at 50X, it is run as:
This hard-masks with "X". To soft-mask (changing basepairs to upper-case except those occurring more than 50 times which are lower-cased.) one can use the commandline option -m SOFT.
The resulting file: "rice.masked.50.fasta" has the same number of chromosomes, each with the same number of basepairs, but with repetitive regions masked. To show the difference, I re-ran the full-genome blast, except this time on the masked genome:
In order to do the full-genome mask, mask_genome.py stores an array for each chromosome, with a length equal to that of the chromosome in a pytables/hdf5 structure which acts like a numpy array. So to increment the counts for a blast hit, the code looks like:
where cache is the hd5 structure (except for some cases it's read into memory to improve speed), 'qchr' and 'schr' are the query and subject chromosome keys with the values being the count arrays, and 's/qstart', 's/qstop' are the bounds of the subject and query for a particular blast line. This sends much of the work to numpy, instead of iterating over the range of qstart, qstop in python. That is repeated for every row in the blast file.
Once all of the counts are created, the script uses that data to mask out the original sequence for any element in count that is greater than the cutoff. That is achieved in a single line iterated per chromosome:
where `np` is from "import numpy as np", `seq` is the original fasta sequence in upper case, `mask_value is either a constant, like 'X', or in the case of soft-masking, a lower-case copy of `seq`, and `hit_counts` is the array of counts. The only tricky bit there is the use of numexpr which makes things a bit faster. The `masked_seq` is written to file after it's corresponding header, and the masking is performed for the next chromosome. The entire masking takes under 10 minutes on my machine.
Generally, we used the masked fasta file as the final output, but there's useful information in the hd5f file--a sort of measure of repetitiveness at each basepair in the genome. also in the svn repo is a short matplotlib web script (for mod_wsgi/mod_python but can be modified to run as cgi or command-line) to generate an image given a request like:
where 'org' is the name of the organism sent to the mask_genome.py script, `xmin`, `xmax` specify the extents in basepairs, and `seqid` the chromosome.
Here's an example from our genome viewer (built on openlayers) where I've added copy count as a layer (with admittedly poor cartography). The nice gene renderings and basepair location ruler are courtesy of Eric Lyons.
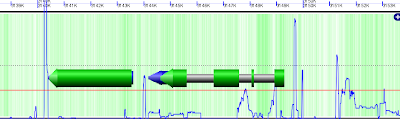 The height of the blue line indicates the copy count. The flat red line is at a copy count of 50. So, the genes in this region are repetitive, but it's possible to see that they are surrounded by long terminal repeats, and that there is some repetitive sequence in the introns (the gray between the big green exons). Scrolling along, openlayers-style, it's possible to see patterns, and spot transposons.
The height of the blue line indicates the copy count. The flat red line is at a copy count of 50. So, the genes in this region are repetitive, but it's possible to see that they are surrounded by long terminal repeats, and that there is some repetitive sequence in the introns (the gray between the big green exons). Scrolling along, openlayers-style, it's possible to see patterns, and spot transposons.
I've made a few genomes available and will be adding more. There's also a (currently undocumented) service to plot the genome. So if the organism is brachy and the chromosome of interest is Bd1 then:
will result in:

where the image is described above.
If you're doing full-genome analyses, give it a try and let me know any suggestions. There's a full example starting from only a genomic fasta, including the BLAST command in the README.rst included in svn. If you get a genome that's too big to run through a full-genome BLAST (maize, ahem). You can split with "pyfasta -split -n 4", run 4 blasts, concat the results, and send through mask_genome.py.
rice (Oryza Sativa) version 5 sequence looks something like below when run through pyfasta info.
rice.fasta
==========
>1 length:43596771
>3 length:36345490
>2 length:35925388
>4 length:35244269
>6 length:31246789
>5 length:29874162
>7 length:29688601
>11 length:28462103
>8 length:28309179
>12 length:27497214
>9 length:23011239
>10 length:22876596
372.078M basepairs in 12 sequences
So, it's not huge (still only 1/10th the size of human) but, it can be difficult to deal with the entire genome because of the large amount of repetitive sequence and transposable elements. This is sometimes mistakenly referred to as "junk DNA", while that's not true, it does make whole-genome analyses a pain as a the output is dominated by repetitive sequences matching their own families. Doing a blast of the rice genome with this command:
/usr/bin/blastall -K 100 -a 8 -d rice.fasta -e 0.001 -i rice.fasta -m 8 -o rice_rice.blast -p blastn
nearly causes my machine to run out of memory (16G), takes a couple of days to run, and results in a blast output of 5.1G and 84 million rows--that's 84 million blast hits with an e-value below 0.001! By definition, that output is dominated by the repetitive elements. Repetitive elements are interesting, but in the case were we want to look at synteny, we have to wade through that 5.1G of stuff to find the very small chunk of data we need. This adds time to run the sequence comparison, time to parse, time to plot, time to analyze, and data to store, etc...
The solution we use in our lab is to create a "masked" sequence where we 'X' out any DNA sequence occurring more than a given number of times in the original blast. So, from the output of the blast above, any basepair that is covered more than 50 times is hard-masked with 'X'. The single script to run this is here. Available from svn via:
svn checkout http://bpbio.googlecode.com/svn/trunk/scripts/mask_genome
when run as a script with no options, it will print some help text. To mask rice at 50X, it is run as:
python mask_genome.py -b rice_rice.blast -f rice.fasta -o rice -c 50 -m X
This hard-masks with "X". To soft-mask (changing basepairs to upper-case except those occurring more than 50 times which are lower-cased.) one can use the commandline option -m SOFT.
The resulting file: "rice.masked.50.fasta" has the same number of chromosomes, each with the same number of basepairs, but with repetitive regions masked. To show the difference, I re-ran the full-genome blast, except this time on the masked genome:
/usr/bin/blastall -K 100 -a 8 -d rice.masked.50.fasta -e 0.001 -i rice.masked.50.fasta -m 8 -o ricemasked.blast -p blastn. The resulting blast file is only 128MB and 2 million rows (contrast to 5.1G and 84 million rows) or a blast file that's 2.4% of the original size. This makes things simpler, and makes doing genomic analyses more enjoyable. This is a different tool than the dust filter that comes with BLAST or many others, because it masks based on the global coverage at each location (there are similar tools).
Implementation
Counting
In order to do the full-genome mask, mask_genome.py stores an array for each chromosome, with a length equal to that of the chromosome in a pytables/hdf5 structure which acts like a numpy array. So to increment the counts for a blast hit, the code looks like:
cache[qchr][qstart - 1: qstop] += 1
cache[schr][sstart - 1: sstop] += 1
where cache is the hd5 structure (except for some cases it's read into memory to improve speed), 'qchr' and 'schr' are the query and subject chromosome keys with the values being the count arrays, and 's/qstart', 's/qstop' are the bounds of the subject and query for a particular blast line. This sends much of the work to numpy, instead of iterating over the range of qstart, qstop in python. That is repeated for every row in the blast file.
masking
Once all of the counts are created, the script uses that data to mask out the original sequence for any element in count that is greater than the cutoff. That is achieved in a single line iterated per chromosome:
masked_seq = np.where(numexpr.evaluate("hit_counts > %i" % cutoff)
, mask_value, seq).tostring()where `np` is from "import numpy as np", `seq` is the original fasta sequence in upper case, `mask_value is either a constant, like 'X', or in the case of soft-masking, a lower-case copy of `seq`, and `hit_counts` is the array of counts. The only tricky bit there is the use of numexpr which makes things a bit faster. The `masked_seq` is written to file after it's corresponding header, and the masking is performed for the next chromosome. The entire masking takes under 10 minutes on my machine.
Plotting
Generally, we used the masked fasta file as the final output, but there's useful information in the hd5f file--a sort of measure of repetitiveness at each basepair in the genome. also in the svn repo is a short matplotlib web script (for mod_wsgi/mod_python but can be modified to run as cgi or command-line) to generate an image given a request like:
?org=rice&xmin=12&xmax=90000&width=800&seqid=2
where 'org' is the name of the organism sent to the mask_genome.py script, `xmin`, `xmax` specify the extents in basepairs, and `seqid` the chromosome.
Here's an example from our genome viewer (built on openlayers) where I've added copy count as a layer (with admittedly poor cartography). The nice gene renderings and basepair location ruler are courtesy of Eric Lyons.
 The height of the blue line indicates the copy count. The flat red line is at a copy count of 50. So, the genes in this region are repetitive, but it's possible to see that they are surrounded by long terminal repeats, and that there is some repetitive sequence in the introns (the gray between the big green exons). Scrolling along, openlayers-style, it's possible to see patterns, and spot transposons.
The height of the blue line indicates the copy count. The flat red line is at a copy count of 50. So, the genes in this region are repetitive, but it's possible to see that they are surrounded by long terminal repeats, and that there is some repetitive sequence in the introns (the gray between the big green exons). Scrolling along, openlayers-style, it's possible to see patterns, and spot transposons.web
I've made a few genomes available and will be adding more. There's also a (currently undocumented) service to plot the genome. So if the organism is brachy and the chromosome of interest is Bd1 then:
http://copymask.syntelog.com/plot/brachy/Bd1/?xmin=12&xmax=38345&width=600
will result in:
where the image is described above.
If you're doing full-genome analyses, give it a try and let me know any suggestions. There's a full example starting from only a genomic fasta, including the BLAST command in the README.rst included in svn. If you get a genome that's too big to run through a full-genome BLAST (maize, ahem). You can split with "pyfasta -split -n 4", run 4 blasts, concat the results, and send through mask_genome.py.
Comments