ngs / high-throughput sequencing pipeline
This is the minimal set of preprocessing steps I run on high-throughput sequencing data (mostly from the Illumina sequencers) and then how I prep and view the alignments. If there's something I should add or consider, please let me know.
I'll put it in the form of a shell script that assumes you've got this software installed.
I'll also assume your data is in the FASTQ format. If it's in illumina's qseq format, you can convert to FastQ with this script by sending a list of qseq files as the command-line arguments.
If your data is in color-space, you can just tell bowtie that's the case, but the FASTX stuff below will not apply.
This post will assume we're aligning genomic reads to a reference genome. I may cover bisulfite treated reads and RNA-Seq later, but the initial filtering and visualization will be the same. I also assume you're on *Nix or Mac.
The command:
Here's an example of the nicely formatted and informative FastQC report before quality filtering and trimming (scroll to see the interesting stuff):
from that, we can see that (as with all illumina datasets) quality scores are lower at the 3' end of the read. For this analysis, I'll choose to chop bases with a quality score under 28. In addition to the other information, we can see that there is an Illumina Primer still present on many of the reads, so we'll want to chop that out in the filtering step below.
The first command clips the adaptor sequence from $FASTQ and pipes the output to the 2nd command, fastq_quality_trimmer, which chomps bases with quality less than 28 (-t) then discards and read with a remaining length less than 40 (-l) and sends the output to a .trim file.
The command:
where we can see the quality looks much better and there are no primer sequences remaining.
This took the number of reads from 14,368,483 to 11,579,512 (and many shorter--trimmed--reads in the latter as well).
Then, you can run the actual alignment as:
We've done a lot of experimenting with different values for -m, and can affect your results, but -m 1 seems a common choice in the literature. And it's clearly less sketchy than, for example, -m 100 which would report up to 100 alignments for any read that maps to less than 100 locations in the genome.
You can have a quick look at the alignment stats with:
But you'll probably want to use a "real" viewer such as IGV, MochiView, or Tablet, all of which I have had some sucess with.
Note that you may want to choose another aligner, because bowtie does poorly with indels. Something like GSNAP will be better suited for reads where you have more complex variants.
I welcome any feedback on these methods.
I'll put it in the form of a shell script that assumes you've got this software installed.
I'll also assume your data is in the FASTQ format. If it's in illumina's qseq format, you can convert to FastQ with this script by sending a list of qseq files as the command-line arguments.
If your data is in color-space, you can just tell bowtie that's the case, but the FASTX stuff below will not apply.
This post will assume we're aligning genomic reads to a reference genome. I may cover bisulfite treated reads and RNA-Seq later, but the initial filtering and visualization will be the same. I also assume you're on *Nix or Mac.
Setup
The programs and files will be declared as follows.#programs fastqc=/usr/local/src/fastqc/FastQC/fastqc bowtie_dir=/usr/local/src/bowtie/bowtie-0.12.7/ samtools=/usr/local/src/samtools/samtools #files. (you'll want to change these) FASTQ=/path/to/a.fastq REFERENCE=/path/to/reference.fastaMost of the following will run as-is for any set of reads, you'll only need to change the FASTQ and REFERENCE variables above.
Seeing the Reads
If you have a file with 2GB of reads, you likely can't just read (pun intended) it and get an idea of what's going on -- though I've tried. There are a number of tools to give you a summary of the data including stats such as quality per base, nucleotide frequency, etc. While fastx toolkit will do this for you, I've found fastqc to be the best choice.The command:
$fastqc $FASTQwill write a folder "a_fastqc/" containing the html report in fastqc_report.html
Here's an example of the nicely formatted and informative FastQC report before quality filtering and trimming (scroll to see the interesting stuff):
from that, we can see that (as with all illumina datasets) quality scores are lower at the 3' end of the read. For this analysis, I'll choose to chop bases with a quality score under 28. In addition to the other information, we can see that there is an Illumina Primer still present on many of the reads, so we'll want to chop that out in the filtering step below.
Filtering the Reads
The fastx toolkit is not extremely fast, but it has a simple command-line interface for most common operations I use to filter reads (though it--like similar libraries--is lacking in support for filtering paired-end reads). For this case, we want to trim nucleotides with quality less than 28 from the ends of each read and then remove reads of length 40. In addition, we want to chip Illumina adaptor sequence. This can be done by piping 2 commands together:# the sequence identified by FastQC above:
CLIP=GATCGGAAGAGCGGTTCAGCAGGAATGCCGAGACCGATCTCGTATGCCGTCTTCTGCTTGAAAAAAAAAAAAAAAA
fastx_clipper -a $CLIP -i $FASTQ -M 30 | fastq_quality_trimmer -t 28 -l 40 > ${FASTQ}.trim
both fastx_clipper and fastq_quality_trimmer are provided in the fastx toolkit.The first command clips the adaptor sequence from $FASTQ and pipes the output to the 2nd command, fastq_quality_trimmer, which chomps bases with quality less than 28 (-t) then discards and read with a remaining length less than 40 (-l) and sends the output to a .trim file.
Seeing the filtered Reads
We then want to re-run fastqc on the trimmed, clipped readsThe command:
$fastqc ${FASTQ}.trimand the output looks like:where we can see the quality looks much better and there are no primer sequences remaining.
This took the number of reads from 14,368,483 to 11,579,512 (and many shorter--trimmed--reads in the latter as well).
Analysis
Up to this point, the analysis will have been very similar for RNA-Seq, BS-Seq, and genomic reads, but you'll want to customize your filtering steps. The following will go through a simple alignment using bowtie that assumes you have genomic reads.Aligning
The first step is to build the reference bowtie index:${bowtie_dir}/bowtie-build --quiet $REFERENCE bowtie-indexThat will create the bowtie index for your reference fasta in the bowtie-index/ directory. It can take a while so you may want to download the pre-built indexes from the bowtie website if your organism is available. Then, you can run the actual alignment as:
${bowtie_dir}/bowtie --tryhard --phred64-quals -p 4 -m 1 -S bowtie-index -q ${FASTQ}.trim ${FASTQ}.trim.samwhich tells bowtie to try hard (--tryhard), assume quality scores are the latest schema from illumina (--phred64-quals), use 4 processors (-p 4), discard any reads that map to more than one location the the reference (-m 1), use SAM output format (-S) and then where to find the bowtie index and the reads. The output is sent to ${FASTQ}.trim.sam.We've done a lot of experimenting with different values for -m, and can affect your results, but -m 1 seems a common choice in the literature. And it's clearly less sketchy than, for example, -m 100 which would report up to 100 alignments for any read that maps to less than 100 locations in the genome.
View the Alignment
From there, we want to view the alignment. Most tools can handle SAM formatted files, but will perform better with an indexed bam. To get that, we do:# view with samtools filter out unmapped reads and converted to sorted, indexed bam.
${samtools} view -bu -S -F 0x0004 ${FASTQ}.trim.sam -o ${FASTQ}.trim.unsorted.bam
${samtools} sort ${FASTQ}.trim.unsorted.bam ${FASTQ}.trim
${samtools} index ${FASTQ}.trim.bam
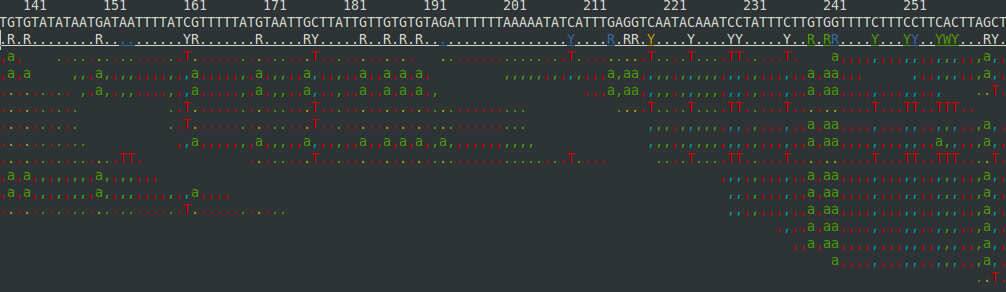
where now ${FASTQ}.trim.bam is sorted and indexed and contains only mapped reads (the .sam file from bowtie contains unmapped reads).You can have a quick look at the alignment stats with:
${samtools} flagstat ${FASTQ}.trim.bamand you can see an awesome ascii curses view of the alignment with:${samtools} tview ${FASTQ}.trim.bamto get something that looks like:But you'll probably want to use a "real" viewer such as IGV, MochiView, or Tablet, all of which I have had some sucess with.
Note that you may want to choose another aligner, because bowtie does poorly with indels. Something like GSNAP will be better suited for reads where you have more complex variants.
I welcome any feedback on these methods.
Comments
Thanks for your post - it's helpful for a newbie like me.
A couple of questions
1. After filtering for quality >28, the data still includes some reads with phred scores below 28. Is this because the Illumina quality score != phred score, or have I missed something?
2. You used a -M 30 param for the clipper method (fastx_clipper -a $CLIP -i $FASTQ -M 30), and -t 28 on the trimmer (fastq_quality_trimmer -t 28) but I don't find this on the hannonlab commandline page. Ummm ... point me in the right direction, please?
thanks
B
feeling my way into bix ...
it will only trim bases that are < 28 and at the end of the read. So if the final base is 30 and 1 base in is 25, it will not be trimmed.
Re: -M 30, you can run the command-line programs with -h to print the help. -M l means require a minimum adaptor length of l.
And -t t gives the quality cutoff.